
Oncolysis + Antitumor&Antiviral Immunity = Success of Oncolytic Virotherapy
Virotherapy is emerging as a new wave of oncological therapies
The approval of tamoligene laherparepvec (HSV-GMCSF, T-VEC, Imlygic®) by FDA and EMA in 2015 has been a milestone, and catalyst for the development of oncolytic viruses (OV). It followed previous approvals of (ECHO-7/Rigvir® in Latvia (2004), and H101/Oncorine® in China (2005), materializing the maturity of several viral platforms.
While ECHO-7 and H101 were the first OV approved worldwide, there is much paucity of evidence that the two drugs actually work. David Gorsky rationally discusses in his blog (from Sept 18, 2017) the concern about the research quality and publications used to establish the credentials of ECHO-7 treatment of melanoma. Similarly, Oncorine (H101; the same construct as ONYX-015), developed by Shanghai Sunway Biotech Co., Ltd since 1999, and approved by Chinese SFDA for nasopharyngeal carcinoma in combination with chemotherapy after the phase III clinical trial, has been halted since 2000 in the United States and Europe due to the lack of efficacy.
Milestones of oncolytic virus therapy development:
1896: First discovery of oncolytic virus
1948-1970: Clinical trials using wilde type oncolytic viruses (Hepatitis B virus, Egypt virus, APC virus, Mumps virus)
1991: Concept of genetically engineered oncolytic virus
1998: Phase I of G2017 for malignant glioma in USA
2005: H101 approved for head and neck cancer in China
2015: T-Vec approved for melanoma in USA
Tumor specificity
The very same mutations that help cancer cells to thrive are targeted by naturally occurring OVs (or those that have serendipitously evolved to become oncolytic; e.g. reovirus and vesic-ular stomatitis virus (VSV)) or genetically engineered OVs (e.g. adenovirus and herpesvirus).
Multiple pathways commonly activated in cancer mitigate cellular antiviral responses (p53, Ras, Rb, Wnt, PTEN, HPV E6/E7, VVEGF, FGF2, ..)
Targeting mechanisms that have been exploited to date can be classified into the four broad categories of transcriptional, translational, transductional and pro-apoptotic.

Figure 1. Mechanisms of tumor targeting by oncolytic viruses. (a) Transcriptional targeting. An essential viral gene is placed under the control of a tumor-specific promoter (some virus promoters are naturally tumor specific). (b) Translational targeting. The virus is engineered (or adapted) to disable viral proteins that antagonize the cellular interferon (IFN) response. (c) Pro-apoptotic targeting. The virus is engineered (or adapted) to disable viral proteins that prevent apoptosis. (d) Transductional targeting. The virus gains entry to its target cells through a receptor expressed more abundantly on tumor cells than on normal cells. Alternatively, the attachment specificity of the virus can be reprogrammed towards tumor antigens by the display of single-chain antibodies or other polypeptide-binding ligands on the viral surface.
The elective susceptibility of malignant cells to OVs stems from cancer-inherent defects in molecular signaling that allow them to evade immune surveillance throughout the oncogenic process. Particularly, many cancers host an impaired type I interferon (IFN) pathway, which compromises their antiviral defenses, and consequently, makes them particularly susceptible to OVs.
Mechanism of action
After infecting cancer cells, OVs hijack the cell death machinery allowing death to occur only after cellular resources have been fully exploited for maximum production of progeny viruses. OVs can kill infected cancer cells in many different ways, ranging from direct virus-mediated cytotoxicity through a variety of cytotoxic immune effector mechanisms. Conventional concepts of cell death—apoptosis, necrosis and autophagy—are generally inadequate to fully describe the complex cell-killing scenarios encountered in virotherapy. This is because the OVs typically takes over and controls the molecular cell death machinery of the infected cancer cell, allowing death to occur only after available cellular resources have been maximally exploited for the synthesis and assembly of new viruses. In addition to direct killing of tumor cells by replication dependent induced cell death, OVs can mediate the killing of uninfected cancer cells by indirect mechanisms such as destruction of tumor blood vessels, promotion of an antitumor response towards all tumor cells (including non-infected cells) by inducing immunogenic cell death (ICD), or through specific activities of transgene-encoded proteins expressed from engineered viruses (Russel et al., 2012).
Recently, the focus in the oncolytic virotherapy field has shifted from their oncolytic effect to their immune stimulatory effect.
Antitumor and antiviral Immunity
In the absence of OVs, antitumor adaptive responses are mitigated by the relative paucity of immune effectors, the presence of immunosuppressive factors and cell types, and poor expression of MHC molecules by malignant cells. The immunological events that follow the administration of OV overturn tumor-associated immunosuppression and promote clinically meaningful antitumor immunity. Potent adjuvant-like immune activities are critical and indispensable aspects of OV-based cancer therapies. Several recent reviews are highlighting the research demonstrating the critical importance of immunity in the overall therapeutic efficacy of OVs (Chaurasiya et al., 2018; Gujar et al., 2018).

Figure 2. OVs as immunotherapeutics. (Chaurasiya et al., 2018)
OV therapies initiate two distinct immunities: antiviral and antitumor. While antitumor immunity is beneficial, antiviral immune responses, of both innate and adaptive nature, have been considered detrimental for the efficacy of OV-based therapy. Indeed, it appears plausible that antiviral immune responses can restrict viral replication and spread, and thus reduce direct oncolysis of cancer cells (and subsequently diminish the efficacy of OV therapies). Considering this, the existing dogma postulates that anti-OV immune responses restrict viral replication and spread, and thus reduce direct OV-mediated killing of cancer cells. Accordingly, a myriad of therapeutic strategies aimed at mitigating anti-OV immune responses is presently being tested.
The innate immunity serves as a first line of defense against viruses, which limits the amplification and spread of viruses, whereas the adaptive immunity plays a major role against the virus during re-infection. Antibodies could potentially neutralize OVs, greatly reducing the virus dose at the tumor site. This may be a concern especially when delivering OVs systemically.
However, the notion that OV-induced antiviral immune responses are detrimental toward anticancer benefits driven by OVs is now facing serious challenges. It's been postulated that antiviral immune responses hold intrinsic anti-cancer benefits and are essential for establishing clinically desired anti-tumor immunity (Gujar et al., 2018). The capacity of antiviral immune responses in targeting OV-infected tumor cells, especially those which remain relatively less susceptible to direct oncolysis, represents a tantalizing therapeutic modality. Thus, the efficacy of OV therapies will ultimately depend on balancing the effects of antiviral immunity between beneficial cancer killing and detrimental restriction of viral oncolysis.
Antiviral immune responses have many antitumor benefits:
(i) adjuvant-like properties of antiviral innate responses are absolutely critical for the initial priming of antitumor immune responses (sensing of PAMPs/MAMPs through one or several of the pathogen recognition receptors (PRRs)),
(ii) intratumoral antiviral immunological events turn ‘cold’ tumors ‘hot’ by facilitating the recruitment of immune cells,
(iii) antiviral immune responses overturn tumor-associated immunosuppression and establish a niche suitable for the development of antitumor immunity,
(iv) OV-mediated oncolysis of cancer cells generates the characteristic signals of immunogenic cell death (ICD) that are pivotal in developing tumor-specific immune attack (types of ICD, such as immunogenic apoptosis, necrosis and autophagic cell death, are characterized by the release of TAAs in combination with DAMPs and viral pathogen associated molecular patterns (PAMPs)),
(v) antiviral CD4+ helper T cells have the capacity to shape the quality of antitumor CD4+ and CD8+ T-cell immune responses (in the absence of OVs, antitumor adaptive responses are mitigated by the relative paucity of immune effectors, the presence of immunosuppressive factors and cell types, and poor expression of MHC molecules by malignant cells), and
(vi) antiviral responses targeting virus replication sites also directly target cancer cells since OVs preferentially infect cancer cells. Beacuse OV-infected cancer cells process and present virus-specific antigens on their surface, antiviral T cells essentially target OV-infected cancer cells.
Antigens and antigen presentation: Viruses can be taken up by antigen presenting cells directly or indirectly when OV-infected cells are engulfed. In the latter case, epitopes are either derived from the virus or from tumor cells. They define the specificity of the T-cell response by designating first infected and later uninfected malignant cells as targets of the host adaptive immune system. OVs enhance the transcription of molecules involved in antigen processing and presentation, such as MHC class I, TAP-1/2, and b2M, in cancer cells, and increase the detectability of cancer cells by tumor-specific T cells.
NK cells: NK cells isolated from OV-treated cancer-bearing mice show enhanced anticancer activities as compared with those from the respective nontreated controls, demonstrating the ability of OVs to reinstate the functionality of tumor-associated innate immune cells. Although NK cells kill infected cancer cells and limit the amplification of OVs, studies have found that NK cells often have positive effects on therapeutic outcomes of OVs.
APCs: DCs, either immature or tumor-associated, achieve maturation following exposure to OVs in vitro and in vivo. Furthermore, OV-mediated killing of cancer cells releases otherwise inaccessible tumor antigens that are captured by APCs, processed in the context of immunostimulatory cytokines, and then presented on their surface in the antigenbinding grooves of MHC molecules.
CTLs: OVs comprehensively overturn tumor-associated immunosuppression and induce tumor-specific CD8+ T-cell (cytotoxic lymphocytes, CTLs) responses that can protect against subsequent rechallenge in an antigen-specific manner. Once developed, these antitumor CD8+ T-cell responses can persist even after OV clearing from the tumor tissue and can actually destroy newly grafted tumors in an antigen-specific but OV-independent manner.
Antibodies: ‘Antibody-mediated complement-dependent cancer cell lysis’ has been shownt to be an important mechanism for therapeutic efficacy of OV both in an animal model as well as in humans (Kim et al., 2013).
Interestingly, Ricca et al. (2017) demonstrate that pre-existing immunity to Newcastle Disease Virus (NDV) may increase its therapeutic efficacy through potentiation of systemic anti-tumor immunity, which would provide clinical rationale for repeated therapeutic dosing. While pre-existing immunity to NDV limited its replication in tumors, tumor clearance, abscopal anti-tumor immune effects, and survival were not compromised and, on the contrary, were superior in NDV-immunized mice.
OVs with immune modulatory transgene(s).
T-VEC (HSV-1 expressing GM-CSF) is the first armed OV approved by the FDA.
Recombinant OVs armed with immune modulators can further enhance the activation of the immune system and overcome the immunosuppressive TME. By introducing
immune stimulators, checkpoint inhibitors and cytokines as immune modulators in viral vectors, adverse events can be reduced, resistance reverted and treatment responsiveness enhanced.

Figure 3. Modulation of the tumor microenvironment by OVs and elicitation of anti-tumor immunity. (i) OVs induce inflammation and increase proinflammatory cytokines (IL-6 and IL-8) and foster tumor infiltration by NK cells and other TILs. (ii) OV infection increases NK cell-mediated tumor cell killing (reduction in MHC I). (iii) Oncolysis by OVs causes ICD with the release of tumor-associated/specific antigens. (iv) Antigen-loaded APCs migrate to the lymph node, where (v) they cross-present tumor antigens to CD8+ T cells. (vi) Following activation, the tumor-specific CD8+ T cells undergo expansion. (vii) The tumor-specific T cells move to both OV injected and un-injected tumors (distant metastases) where they can exert anti-tumor effect. (Chaurasiya et al., 2018)
OVs with immune modulatory transgene(s).
T-VEC (HSV-1 expressing GM-CSF) is the first armed OV approved by the FDA.
Recombinant OVs armed with immune modulators can further enhance the activation of the immune system and overcome the immunosuppressive TME. By introducing
immune stimulators, checkpoint inhibitors and cytokines as immune modulators in viral vectors, adverse events can be reduced, resistance reverted and treatment responsiveness enhanced.

Table 1. Combination therapy of armed oncolytic viruses and immune modulators. (Graaf et al., 2018)
The overall effect of the oncolytic immune therapy depends on the interplay between virus, immune modulator and tumor. On one end OVs armed with immune modulators
induce localized (often more effective, but not necessarily the case for immune checkpoint blockers, in particularly anti-CTLA-4 Ab) concentrations of the modulator compared to systemic monotherapy, however vectorization can also limit the potency of the immune therapies by incorrect localization and timing, and no resilience in case of tumor resistance.
Besides, depending on replication, oncolytic activity and immunogenicity of OV, as well as the insertion site and the size of the immune modulator, its expression may differ substantially.
OVs in the era of immune checkpoint blockers (ICBs)
Recently, Ribas et al. reported the findings of a phase Ib trial in which they studied the impact of T-VEC on therapeutic efficacy of anti-PD-1 antibody pembrolizumab in patients with metastatic melanoma (Ribas et al., 2017). The combination treatment was well tolerated; T-VEC was found to promote T cell infiltration into tumors and improved the overall therapeutic efficacy of pembrolizumab. Chesney et al. (2017) have published the data from randomized open-label phase II study of T-VEC with Ipilimumab in unresectable advanced melanoma. The ORR was significantly higher with T-VEC+Ipilimumab (39%) vs Ipilimumab alone (18%)(2.9; 95% CI, 1.5 to 5.5; P = .002). Responses were not limited to injected lesions; visceral lesion decreases were observed in 52% of patients in the combination arm and 23% of patients in the ipilimumab arm. These data indicate that the combination has greater antitumor activity without additional safety concerns versus ipilimumThese clinical trial essentially confirmed the findings from animal studies (see below) that OVs can enhance the therapeutic efficacies of ICBs by converting immunologically ‘cold’ tumors into immunologically ‘hot’ tumors. These trials provides the hope that the benefits of checkpoint inhibitors may be harnessed in combination with OVs, even in tumor types that have previously shown very poor response to checkpoint inhibitors, such as breast, prostate and colon cancer.
Interestingly, OVs themselves promote the expression of checkpoint molecules on the surface of cancer cells. Therefore, both therapeutics seem to complement each other and the outcome is a synergistic anti-tumor effect.
The strategic administration of oncolytic adenovirus, NSV, measles or vaccinia virus in combination with checkpoint inhibitors has produced better therapeutic efficacy as compared with either monotherapy - see the side bar Zamarin et al. (2018) and below.
In 2015 Woller et al. showed that localized tumor infection with an oncolytic adenovirus could overcomes systemic tumor resistance to PD-1 inhibitor by broadening neoantigens-directed T cell responses in mice. Oncolysis of the primary tumor significantly abrogated systemic resistance to PD-1-immunotherapy leading to improved elimination of disseminated lung tumors.
Bourgeois-Daigneault et al. (2018) discovered a way to address the ICB resistance for breast cancer, and Samson et al. (2018) discovered a way to address this for brain tumors. In both cases, the authors found that oncolytic virus treatment given early, before surgical resection, alters the antitumor immune response and potentiates the effects of subsequent treatment with immune checkpoint inhibitors. Although these studies differ in the details of their methods and the immune effects induced by the oncolytic viruses, they indicate the potential of such viruses for enhancing the potential of checkpoint therapy and expanding it to new types of cancer (also see side bar).
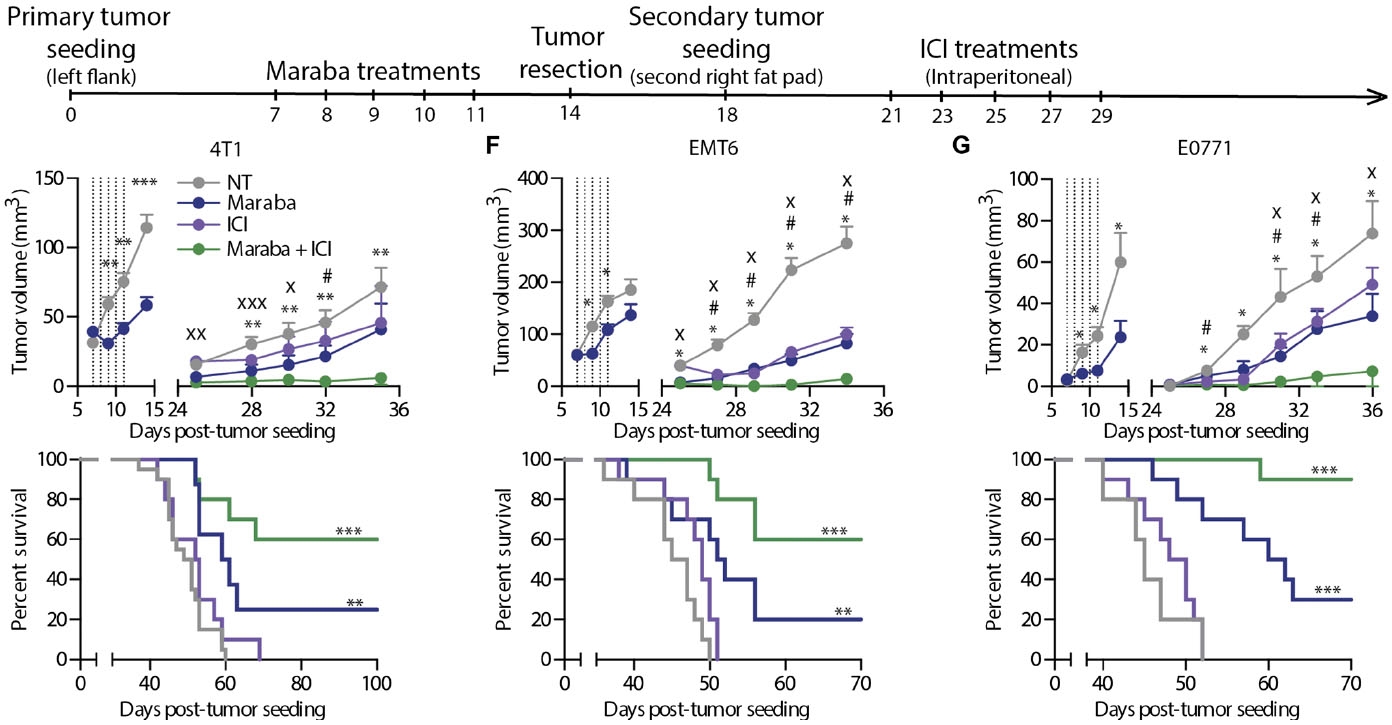
Figure 4. Maraba treatment results in complete responses in the window of opportunity setting. (A) Schematic representation of the treatment schedule used for the tumor rechallenge model. (B to D) Tumor growth and Kaplan-Meier survival curves obtained using 4T1 (B), EMT6 (C), or E0771 (D) cells in the tumor rechallenge model (n = 10 mice per group per experiment). Maraba treatments were administered intratumorally. NT, no treatment. (E and F) The same experiment as in (B) was repeated using intravenous delivery of Maraba virus (E) or immunocompromised CD-1 nude mice (F). (G) Primary EMT6 or 4T1 tumors were treated with Maraba or left untreated and were resected, and all animals were rechallenged with 4T1 tumors. The dotted lines indicate the time of Maraba treatment. Statistical analysis for tumor measurements: *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired multiple two-tailed t test). Statistical analysis for survival curves: *P < 0.05, **P < 0.01, ***P < 0.001 (Mantel-Cox test). (Bourgeois-Daigneault etal., 2018)
Although the response data coming from the phase Ib and II studies are highly promising, we need to await the results of the ongoing randomized phase III trial to properly understand the potential synergy between local administration of a viral vaccine and PD-1 blockade. In addition, such a larger study will also be of value to address another key question that is raised by the current work. is which subpopulation of patients may benefit most from combination therapy over single-agent PD-1 blockade? Is this the group of patients with highly mutagenized tumors that failed to attract T cells, or is combination therapy lowering the average mutational burden that is required for clinical response? If the latter, implications for the many other tumor types with lower mutational burdens could also be profound (Haanen et al., 2017).



