
Guidelines for the next-generation cancer vaccines
Immuno-oncology field is experiencing a harsh realization that the applicability (i.e. benefit) of immune checkpoint blockers (ICB) is restricted to only a subset of patients. See previous blog (2018 I-O Research challenges) on the current directions in approaching the challenges in cancer immunotherapy. There are several limiting factors underlying the non-responsiveness to ICB therapy, including low tumoral T cell infiltrate (so called 'cold tumor phenotype') and low neoantigens burden.

Figure 1. ORRs-based (arbitrary) comparison of efficacy of conventional chemotherapy/radiotherapy, targeted therapy, cytokine therapy, DC vaccines, and specific ICIs against melanoma, glioblastoma, and renal cell carcinoma (RCC). (Garg et al., 2017)
A rational approach warrants for a combinatiorial therapy ensuring that the host immune system is pre-sensitized to the tumor. Apart from amplifying the existing responses, cancer vaccines have the potential to generate new antigen-specific T cell responses and thereby fill the void of the current therapeutic modalities aimed at restoring the exhausted phenotype of existing anti-tumor CTLs.
Therapeutic cancer vaccines have been extensively tested in patients with advanced cancer but have had little clinical success and have greatly under-represented the full potential of the vaccines. Nevertheless, with the increasing knowledge gained from the immunological and clinical studies we are now getting to understand the fundamental issues of the past vaccine-based immunotherapy approaches. Furthermore, recent technological advances in systematic discovery of tumor neoantigens enabled the emergence of personalized cancer vaccination as a novel promising treatment strategy.

Figure 2. History of cancer vaccines (Hu et al., 2017)
1. Fundamentals of anti-tumor vaccines
The ultimate goal of anti-tumor vaccines, i.e. productive activation and tumor-infiltration of tumor-specific T cells requires delivery of 4 separate signals during antigen-presenting cell (APC) - T cell interaction:
- a cognate antigen, called signal 1, [the most prominent APCs are dendritic cells (DCs), since they can accomplish both conventional presentation (i.e. present endogenous antigens as MHC I - associated peptides to CD8+ T cells) and cross-presentation (i.e. presenting exogenously captured antigens as MHC I-associated peptides)]
- costimulatory cues from ligands present on the APC surface (e.g., CD80, CD86, CD40; signal 2),
- proinflammatory cytokines (e.g. IL-12), which constitutes signal 3 and
- the induction of tumor homing properties of tumor-specific T cells (e.g. IL-12, vitamin A, vitamin D; delivery of signal 4)
Contemporary studies have established a defining role for the dying cells as a further upstream point of regulation. Dead cells were shown to be a dominant source of DAMPs (released upon cellular damage and accidental necrosis), thus providing signal 0 and antigen for CD8+ T cell cross-priming. Different mechanistic forms of cell death may influence the outcome of DC-mediated cross-presentation. The activation of cell death pathways is an initiating immunological event resulting in the generation of signal −1. This signal includes constitutive DAMPs (cDAMPs), which are immune-stimulatory cellular factors that are present before death and are released by dying cells, and inducible DAMPs (iDAMPs), which are factors that are actively produced or modified during cell death (reviewed by Yatim et al., 2017).

Figure 3. The initiation of adaptive immunity by dying cells requires a set of immunological events, the coordination of which leads to the priming of T cells. Four dendritic cell (DC)-derived signals act on T cells (lower panel): signal 1 is the antigen-recognition event that is mediated through the T cell receptor (TCR), and triggered by MHC class I-associated or MHC class II-associated peptides processed from the antigen after the phagocytosis of dying cells. Signal 2 is the co-stimulation event, which is mediated by engagement of CD28 by CD80 and CD86. Signal 3 is the polarizing and differentiation signal delivered from the DC to the T cell that determines its differentiation into an effector cell. Soluble molecules such as interleukin-12 (IL-12) family members, and interferon-α (IFNα) and IFNβ, are mediators that deliver signal 3. There are data indicating that DCs may also provide T-cells with an additional signal (tentatively termed 'signal 4'), which regulates organ-specific trafficking of immune cells. The upregulation of co-stimulatory molecules (signal 2) and the secretion of inflammatory cytokines by DCs (signal 3) depend on the activation of pattern recognition receptors (PRRs) by microbe-associated molecular patterns (MAMPs), or by constitutive or inducible damage-associated molecular patterns (cDAMPs or iDAMPs, respectively; lower left panel). The DC activation event has been termed signal 0. Recent data have established that the activation of innate immune pathways within dying cells, such as the nuclear factor-κB (NF-κB) pathway, represents an earlier immunological event that regulates the outcome of T cell priming. We propose to designate this event signal −1 (upper panel), although the manner by which it influences the immune response is still unclear. IRF, IFN-regulatory factor. The dashed arrow indicates crosstalk between cell death effectors and innate immune pathways. (Yatim et al., 2017)
Failure to concommitantly deliver all signals to T cells fails to ensure effective type-1 immune polarization and induction of cytolytic CD8+ T cells (CTLs), critical effector cells for anti-tumor immunity.
Tumors find one way or the other to prevent the delivery of these signals by either the loss of antigen or direct induction of DCs dysfunction, and further suppress anti-tumor immunity by suppressing CTLs via the production of immunosuppressive cytokines, expression of IC, lack of CTL-attracting chemokines, ...
Any agent capable of arming DC cells to present these signals can be considered as anti-tumor vaccine, bypassing the tumor's inherent and TME-induced obstacles abrogating effective tumor-antigen presentation. While whole tumor cell vaccines and antigen-delivery vehicles (recombinant/synthetic antigenic-peptides) may serve as the source of antigens to be taken up by DCs for presentation, they do not provide the relevant signals for DC activation and therefore do not integrate several immunologically relevant signals required for the efficient induction of antigen-directed T-cell responses.
Two key components of the vaccines are therefore: antigenicity and immunogenicity
Tumor antigens
Antigens exploited in cancer immunotherapy are derived either from non-mutated self-proteins which are differentialy expressed between normal and tumor cells, so called - tumor associated antigens (TAA) - overexpressed antigens, differentiation-specific antigens and cancer-testis antigens (CTA) or
- from mutated proteins and therefore tumor-specific antigens, comprising products of oncogenic viruses (HBV, HPV, Epstein Barr virus), oncogenes, tumor suppressor genes and neo-antigens (generated as products of somatic mutations) (see Figure 1).

Figure 4. Important tumor antigens for vaccines (Finn, 2017)
For overexpressed and tissue differentiation antigens the anti-tumor immune response is presumably induced by high levels of antigen expression, which breaks the immunological tolerance, while CTAs are thought to provide higher tumor specificity, as they are not expressed in normal tissues (excluded germline and trophoblastic cells). Natural T cell recognition of TAA is often of low affinity as a result of negative selection of high-affinity clones during T cell development.
Because oncogenic viral antigens and neo-antigens are foreign to the body (and therefore not subject to central tolerance) and expressed only by cancer cells (and therefore specific for the tumour), they are highly suitable for use in a cancer vaccine.
Immunogenicity
Tumour neoantigens are generated as products of somatic mutations, and hence they are not only exquisitely tumour specific but also highly immunogenic on the basis of lack of central tolerance. The evidence supporting their immunogenicity (Tran et al., 2015; Linnemann et al., 2015) and their use for developing personalized vaccines:
- higher neo-antigen load is associated with stronger T cell responses and better clinical outcome (including clinical response to CPB therapy) (Brown et al., 2014; Van Allen et al., 2015)
- neo-antigen specific T cell populations are expanded in the setting of effective antitumor immunity (Van Rooij et al., 2013)
- neo-antigen specific T cells are cytolytic for tumor cells presenting mutated peptides and can contribute to complete or partial tumor regression (DuPage et al., 2012)
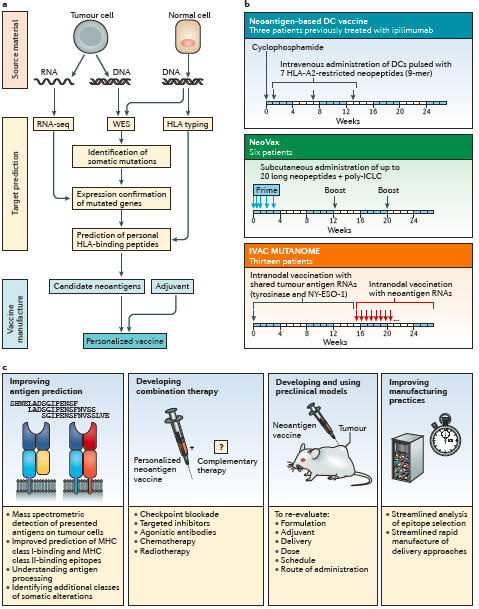
Figure 5. Neoantigen-based therapeutic cancer vaccines. a | The typical workflow for neoepitope selection and vaccine manufacture. b | The schema of three phase I clinical trials of personalized neoantigen vaccines in patients with melanoma. c | Strategies to improve personalized neoantigen vaccines for cancer (Hu et al., 2017)
While tumor antigens harbor a specific level of inherent immunogenicity (associated with antigenicity), as mentioned above, in the absence of inflammatory stimuli presented antigens induce tolerance rather than immunity.
Effective immunization strategies therefore require either preconditioning (by eliciting local inflammatory response at the vaccine site with a recall antigen such as tetanus-diphteria toxoid), co-administration of an immune adjuvant (TLR agonist, CD40 agonist, GM-CSF, STING agonist, ..) and/or inducing immunogenic cell death (ICD).
Tumor cell death
In case elicited in an immunogenic manner, tumor cell death may function as an inherent vaccine. Alongside passive signals that are generated by dying cells, the upstream immunological cues may actively regulate adaptive immunity.
Critical determinant of immunogenic cell death is the antigen cross-presentation. Although proteasome activity within APCs is undoubtedly important for the processing of internalized intact protein, antigen-donor cell proteasomes have also been shown to process antigen and the generated peptides that can be directly loaded onto DC MHC class I molecules. Caspase activation has the ability to expose neo-epitopes through the direct cleavage of proteins, which results in epitopes from these proteins being favoured for cross-presentation. E.g. caspase 3- mediated cleavage of proteins in antigen-donor cells is crucial for the priming of self-antigen-specific CD8+ T cells. Two factors that have an important role in successful cross-presentation are overall antigen expression level and antigen stability.
The second determinant is immunogenic stimuli. Germline-encoded signals can also trigger immunity, meaning that the endogenous and constitutively expressed molecules released from damaged tissue can be sensed by appropriate receptors on DCs, which in turn can stimulate adaptive immune responses. Therefore active mechanisms, occurring alongside programmed cell death, might contribute to the immunogenicity of dying cells. While the key–lock paradigm proposed that the exposure or release of three different DAMPs — calreticulin, HMGB1 and ATP — by apoptotic cells accounts for the immunogenicity of dying cell (Zitvogel et al., 2010), since, the data demonstrated that the release of endogenous DAMPs such as ATP and HMGB1 is insufficient, and that NF-κB activation within dying cells is a key determinant for achieving T cell priming (Yatim et al., 2015). Coupled activation of innate immune pathways and cell death pathways within the antigen-donor cell regulates the outcome of antigen cross-presentation.
[Importantly, inflammatory cell death and immunogenic cell death should not be conflated, meaning that the stimulation of DC maturation or the provocation of an inflammatory response will not necessarily translate into the cross-priming of CD8+ T cells. ]
Recently, therefore DAMPs have been classified into cDAMPs (HGMB1, uric acid crystals, histones), which are preformed molecules, and iDAMPs, which include molecules that are generated actively during the process of cell death via mechanisms that are dependent on the underlying cell death pathway (chaperones HSP70, HSP90, type-1 interferons, IL1b, IL18, ..). cDAMPs are well validated as mediators of inflammation, stimulate PRRs on the surface of DCs and provide signal 0, thereby initiating DC migration to the lymph nodes, however they are inefficient in CD8+ T cell priming in the absence of NFκB activation. iDAMPs also signal innate immunity (for example, activation of receptor-interacting protein kinase 1 and nuclear factor-κB) in the antigen-donor cell as a result of cell stress and death. The activation of innate immunity pathways within dying cells is an initiating immunological signal −1 (Yatim et al., 2017).
2. Where did the past vaccination strategies fail ?
Initial prioritization of tumor antigens for vaccination considered the available evidence
of their expression, relative antigenicity, and immunogenicity, with the lack of awareness about the possibilites presented by the use of neo-antigens. The first clinical trials either tested peptide-based vaccines using TAAs, or tested whole tumor cells or cell lysates that contained some of the known TAAs. There are two major caveats in targeting non-mutated self-antigens. First, the immunogenicity of a self antigen is dampened because of reduction in the number of high-affinity self-reactive T cells, which result from tolerating mechanisms such as clonal deletion, ignorance, anergy, or suppression in the host. Meaning that dominant TAA-derived peptides, although potentially immunogenic because of their high HLA-I affinity, are not highly immunogenic because the specific T cells repertoire has been rendered tolerant. This explains why all cancer vaccines tested in clinical studies to date failed to show clinical benefit- all of the vaccines targeted TAA derived dominant peptides. Second, apart from the continual deletion or tolerization of self-reactive T cells against dominant epitopes, the poor immunogenicity of low-affinity ('cryptic') self/tumor antigens is due to the instability of the peptide-MHC complex, which cannot break. While the T cell repertoire specific for the TAA-derived low-affinity peptides is fully available to be mobilized, the stability of the pMHC-TCR complexes limits their immunogenicity (see below the optimization of peptide-MHC binding with heteroclitic peptides). Further, without concommitant activation provided by adjuvants, such efforts precluded proper T cell licencing to kill.
Therefore, despite the presence of surprising widespread T cell recognition of self antigens on human cancer cells, understanding the critical role of stable peptide-MHC complex in immunological synapse formation explains why vaccination strategies using self-antigens (and further in the absence of proper adjuvants) will very unlikely trigger the treshold for an effective stimulation of low-affinity T cells in cancer patients.
With the discovery and ascension of DCs (1975), they became a prominent delivery vehicle for TAAs. While first -generation DCs consisted of either patient-isolated natural DCs or ex vivo-generated monocyte-derived immature DCs (mo-iDCs), the second-generation DCs were fully matured, yet the maturation cocktails often lacked the type-1-driving capacity (i.e. induction of IL-12 production by DCs) for induction of effective CTLs. This most likely being the major reason behind the scarcity of past DC vaccine success.
Extensive empirical clinical experience has suggested that vaccines targeting single tumor antigens are inadequate for addressing tumor heterogeneity and for meeting the challenge of clonal evolution and immune escape by the tumor.
Altogether, reasons for dissapointing therapeutic vaccination attempts include not only immune evasion upon selection of antigen loss variants but also weak vaccine formulations and a limited, low-avidity and tolerized T cell repertoire that is deployed in an immunsuppressive tumor environment. Not to mention that none of these early vaccination efforts were combined with ICB, therefore any effective anti-tumor immunity that these vaccines induced encountered the resistance from upregulation of immune checkpoints and loss of sensitivity to intereferons (IFNs).
3. Shaping the next generation vaccines
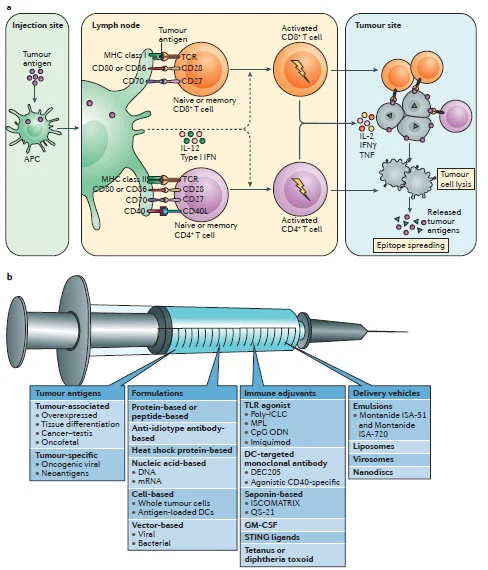
Figure 6. Mechanisms and components of an effective cancer vaccine. a | The tumour antigen presentation process. b | There are four key components of cancer vaccines: tumour antigens, formulations,
immune adjuvants and delivery vehicles (Hu et al., 2017).
Antigen selection: Is there an off-the-shelf alternative to neo-antigens ?
With the recent advances in the field (see the side column about (Ott et al., 2017; Sahin et al., 2017)), it has become evident the tumor antigen prioritization needs to be revisited. Informed prioritization of tumor antigen candidates
for vaccine development should consider the degree of relatedness to self-antigens. With
such a metric, virally encoded antigens and neoantigens would be at the top of the list. Further, frequently mutated sequences in some tumors (such as the epidermal growth factor receptor vIII isoform in glioma) would also represent important targets for vaccination strategies. Posttranslationally modified peptides and CT-type antigens, differentiation antigens and overexpressed antigens should follow (the antigen prioritization outlined by Romero et al., 2016).
Tumor-specific mutated oncoproteins represent the ideal antigens because they are not only highly tumor-specific and therefore antigenic, but also provide a distinct level of predictability. There is an expanding spectrum of identified driver alterations offering tremendous opportunity for such therapeutic strategy. The most critical issue that needs to be taken into account for vaccines harboring oncoproteins is tumor heterogeneity and antigen loss (through epigenetic or post-translational means). Simultanous targeting of several mutated oncogene-encoded antigens would be the best mode to avoid such tumor-evolved resistance mechanisms.

Figure 7. Multiple novel oncogenic drivers have been identified in non-small-cell lung
cancer (NSCLC) that might be amenable to therapeutic targeting (Rotow et al., 2017).
The posttranslational modifications as an alternative source of neoantigens occur due to dysregulated signaling in cancer. For example, many of the tumor-associated phosphopeptides were shown to be derived from oncogenes, making these of particular interest as immunotherapeutic targets (Cobbold et al., 2013). Peptides bearing mono- and dimethylated arginine residues can be presented for recognition by T cells and may therefore offer a novel target in immunotherpy (Marino et al., 2016).
As discussed above vaccinating against self-antigen (i.e. TAA) with self-antigen is at best far from optimal. With the knowledge gained from the failed vaccination strategies, we need to consider alternatives to inducing an immune response to non-mutated self-antigen. The presence of foreign sequences in the vaccine protein is an essential requirement to circumvent tolerance (Saupe et al., 2015). A strategy that has been applied to immunize against non-mutated TAAs is vaccination with a mutated form of TAA - heteroclitic peptides. As a proof-of-concept for immunotherapeutic potential for heteroclitic peptides, Bae et al., 2014 demonstrated the induction of CTLs with specific antitumor activities against a variety of solid tumors by a cocktail of immunogenic HLA-A2 specific heteroclitic unspliced and spliced XBP1 peptides. Further, in support of the above, Vaxon biotech reported in June 2017 the results of a Phase IIb clinical trial of Vx-001 (a therapeutic vaccine based on optimized heteroclitic peptides) in HLA-A*0201 positive patients with TERT expressing NSCLC (stage IV and distant recurrent stage I-III) and controlled disease after first line chemotherapy (NCT01935154) showing that Vx-001 drastically and very significantly prolonged overall survival (20.7 months for Vx-001 vs 7.9 months for placebo ; p=0.0007) and time to treatment failure (5.6 months for Vx-001 vs 3.1 months for placebo ; p=0.006) in never and light smoker patients with non-immunogenic tumors. They also strongly suggested that Vx-001 can turn tumor initially resistant to immune checkpoint inhibitors sensitive.
The scientific rationale behind this is that TAA peptides often lack canonical anchor residues, which can be substituted for the optimal residue to improve their antigenicity. Immunization with the mutant self peptide having a high affinity for MHC-I molecules compared to self peptide, is capable of overcoming the instability of self peptide MHC-I complexes and activating the T cells which are then competent to recognize and kill tumor cells presenting the non-mutated self peptide (Yu et al., 2004). This is enabled by the enigmatic specificity of TCRs; TCRs are at once specific but also cross-reactive (Singh et al., 2017). The caveat of this approach being the possibility that the T cells primed with these heteroclitic peptides subsequently fail to recognize the natural, tumor-expressed peptide, which may provide a molecular reason for why clinical trials of these peptides have been thus far unsuccessful (Lesterhuis et al., 2011). The essential T-cell cross-reactivity between the natural and modified (heteroclitic) peptides for this approach to work depends on whether the anchor residue substitution influences peptide conformation (Madura et al., 2015). The development of predictors of the TCR/pMHC binding mode, recognition of immunodominant epitope (Song et al., 2017) and the antigen specificity of a given T cell from its TCR sequence will help shape designing the suitable peptide(s) for vaccination.
How about aiming broader
Multipeptide vaccines have shown improved efficacy compared with single-peptide formulations, as they are less likely to trigger immune escape. Long peptides (20-30mer), rather than 8-10 mer (predicted to bind directly to MHC I) require internalization and processing by professional APCs, before MHC-restricted presentation, as well as generally contain MHC II-restricted peptides, therby providing optimal activation of T cells.
Whole tumor vaccines (including whole tumor-loaded DCs) may present an alternative to exploiting neo-antigen(s) and well-designed/predicted mutated versions of TAA as improved next-generation vaccination approaches. Ultimate advantages of this approach are that it obviates challenges in defining specific antigens for vaccine development and offers the benefit of tumour and/or patient-specific antigen presentation to elicit immunity against a broad spectrum of (neo)-antigens.
An important consideration when applying whole tumor/cell aproaches is the relevance of MHC compatibility. Namely, accumulating data indicate that the efficient induction of antigen-specific CTLs is ultimately caused by inherent bystander DCs (reports of a FIH study recently published - Laurell et al., 2017; Fotaki et al., 2017). In such a scenario, administratoin of pre-activated allogeneic DCs producing high levels of inflammatory factors themselves (but also further inducing a local rejection process at the injection site), induces the recruitment and maturation of endogenous bystander DCs, which efficiently cross-present tumor antigen(s).
With this in mind, it seems that all fingers point towards ICD induction as a mode of intrinsic vaccination by using the patient’s own tumor/TME in situ.
Let's not forget the critical role of adjuvant(s)
The significant progress in understanding the molecular mechanisms involved in the induction of immune responses highlighted the potential use of TLR agonists alone or in combination with other adjuvants for vaccines. For example, monophosphoryl lipid A (MPLA), a TLR4 agonist is currently used in human licensed vaccines for the prevention of HPV and HBV infections. Several other adjuvants, including MF59, adjuvant system (AS) 01, AS03, AS04, and virosomes, are now available in licensed vaccine products. In other cases however, the adverse effects prompted efforts to design new molecules with better safety profiles and potent adjuvanticity. Notable examples include analogs, which belong to the families of muramyl dipeptide (MDP), MPLA, TLR2 agonist Pam3CSK4, TLR7 agonist imiquimod, TLR9 ligand CpG oligodeoxynucleotides (ODNs), QS-21, an acylated 3,28-bisdesmodic triterpene glycoside, and a-GalCer. Most of these molecules are pathogen-associated molecular patterns (PAMPs), which can induce immunity by interacting with receptors of the innate immune system, such as TLRs, NOD-like receptors, and RIG-I (a family of cytoplasmic RNA helicases)-like receptors (for comprehensive overview of novel adjuvants designed for improving vaccine efficacy see Bonam et al., 2017). A new class of immune adjuvants being evaluated in preclinical studies and in an early phase clinical trial in patients with advanced/metastatic solid tumors (NCT02675439) are cyclic dinucleotides, which signal via stimulator of interferon genes (STING), a crucial sensor of DNA viruses (for more about STING agonists see previous blog 2018 I-O Research challenges).
A growing recognition of the critical role of immunogenicity for effective vaccination approaches, launched widespread clinical implications for previously neglected delivery vehicles in anti-tumor vaccination. Two such modalities are mRNA vaccines and oncolytic viruses.
The use of mRNA vaccines has several beneficial features: mRNA is a non-infectious, non-integrating platform, and is degraded by normal cellular processes, various modifications make mRNA more stable and highly translatable, and perhaps most importantly, it is inherently immunogenic (Pardi et al. 2017 have just published an extensive review on mRNA vaccines that is worth of in-depth reading).

Figure 8. Leading mRNA vaccine developers: research focus, partners and therapeutic platforms (Pardi et al. 2017).
The ideal oncolytic virus will have multiple therapeutic activities, i.e. oncolysis and anti-tumor immune stimulation. Through the induction of tumor cell lysis, presentation of (neo)-antigens is enabled besides the triggering of local inflammatory response. More on Oncolytic viruses in one of the future blogs (subscribe to the Newsletter).
The necessity of combinatorial strategies - at least this is obvious
Complementary therapies to reverse immune suppression in the tumour microenvironment — such as depleting regulatory cells, inhibiting regulatory molecules or blocking metabolic suppression — will be important to unleash the full potential of a cancer vaccine. Several studies have suggested potential additive or synergistic effects between a cancer vaccine and CPB.
Strategies such as targeting multiple (neo-)epitopes and combining personalized
vaccines with complementary therapies such as CPB will be important to prevent immune escape. For example, anti–CTLA-4 treatment of patients with metastatic melanoma reportedly broadens the T cell responses to shared antigens such as overexpressed antigens, differentiation antigens, and CT antigens.

Figure 9. Integrating next-generation vaccines into highly efficacious, biomarkers-driven, combinatorial regimens against cancer.
Vaccines should be ideally administered in a repetitive manner to achieve maximal long-term effects. The reported data suggest that the prime-boost strategy as a combination of vaccines (i.e., heterologous prime-boost) may be better than a single vaccine, as heterologous prime-boost can be more immunogenic than homologous prime-boost strategy (a review on prime-boost vaccine strategies by Kardani et al., 2016). Further, a heterologous prime-boost protocol ensures that the response against the tumor antigen, the only antigen shared between the two vaccines, is enhanced by the boost. A proof-of-concept data was reported by Aduro at SITC, 2017, showing that a heterologous prime-boost vaccination strategy combining a listeria and DNA-based vaccine encoding prostatic acid phosphatase (PAP) elicits a strong antigen-specific, anti-tumor response.
Future studies will focus on improving antigen prediction, improvement of HLA-binding algorithms alongside with the determination of α/β pairs using large-scale, single-cell analysis to better understand the number of unique TCRs required by an individual to assure immune protection. Such information could provide an opportunity
to increase the likelihood of targeting (neo-)antigens that are expressed by a patient’s cancer cells and potentially be used to decide whether prescription of a vaccine is necessary and whether a protective number and type of T cells are induced post-vaccination.
4. Pre-malignant lesions as targets for preventive vaccines
The remarkable results for HPV vaccines and viral antigens that had not shown previous clinical efficacy in invasive disease suggest that vaccines based on nonviral antigens that failed in the therapeutic setting may also more likely succeed in the prevention setting. While current managment of pre-malignant lesions is either surgical removal or frequent screening, using cancer vaccines in the pre-malignant setting seems most rationale to boost immunosurveillance to help eradicate residual disease and prevent its recurrence and/or progression into cancer with degree of risk that is acceptable when compared to healthy individuals.

Figure 8. The dawn of vaccines for cancer prevention (Finn, 2017)
In contrast to therapeutic indications, personalized preventive vaccine approach (reviewed recently by Finn 2017) is currently highly impractical and resource-demanding for a preventive application. A more practical approach that could yield an off-the-shelf vaccine against mutated antigens would be to use predictable mutations, such as those in the RAS family of oncogenes or the cancer suppressor gene TP53. The result of targeting mutations in oncogenes and cancer suppressor genes might be much different in a preventive setting and these mutations occur very early in the process of tumorigenesis; many pre-malignant lesions, such as those leading to pancreatic, breast, lung or colon cancers, already harbour KRAS or TP53 mutations. Similar to a cancer setting, pre-malignant lesions express TAAs, such as SOX2, MUC1, cyclin B1, .. for which similar strategies to enhance their immunogenicity can be applied as described above.
......................................................................................................................................................................................................................................................................................................................................



